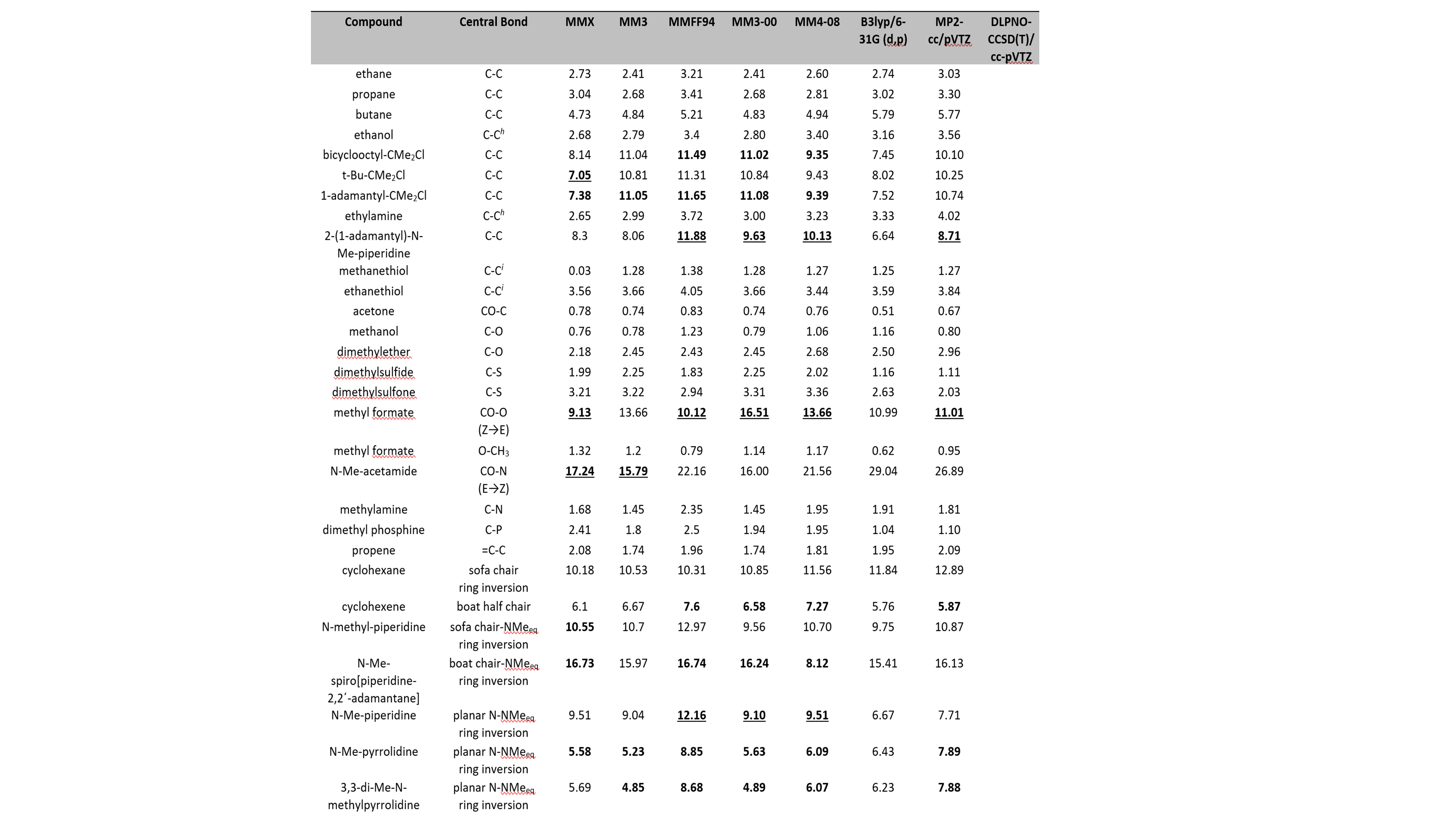
The evaluation of various computational chemistry methods, molecular mechanics calculations, ab-initio QM methods, on their ability to predict the experimental conformational preferences of organic molecules and drugs. Example of a subset of compounds and conformational energies is given in Table 1.
A total of 160 conformational energies from 151 model organic molecules were examined and many conformational update aspects were outlined. The data set of the organic molecules included hydrocarbons, haloalkanes, oxygen containing compounds, nitrogen containing compounds, conjugated compounds, phosphorus and sulphur containing compounds. The study allowed the comparison of some standard force fields with wide distribution in commercial and free software for the calculation of conformational energies. The molecules were energy minimized and a number of conformational energies and barriers for the compounds were calculated using UFF, DREIDING, MM2-91 in the form of MM2, MMX, MMFF94, MM3-96, MM3-00, MM4. The performance might be interesting for the force field used from algorithms to produce conformations before docking.
The energies were also calculated using B3LYP/6-31G(d,p), MP2/6-31G(d,p) and also with very accurate theory DLPNO-CCSD(T)/cc-pVTZ using ORCA software at the limit of the correlation energy which has never been applied (with Professor D. Pantazis, Germany).
Table 1. Relative conformational energies of some intramolecular barriers (kcal mol-1).
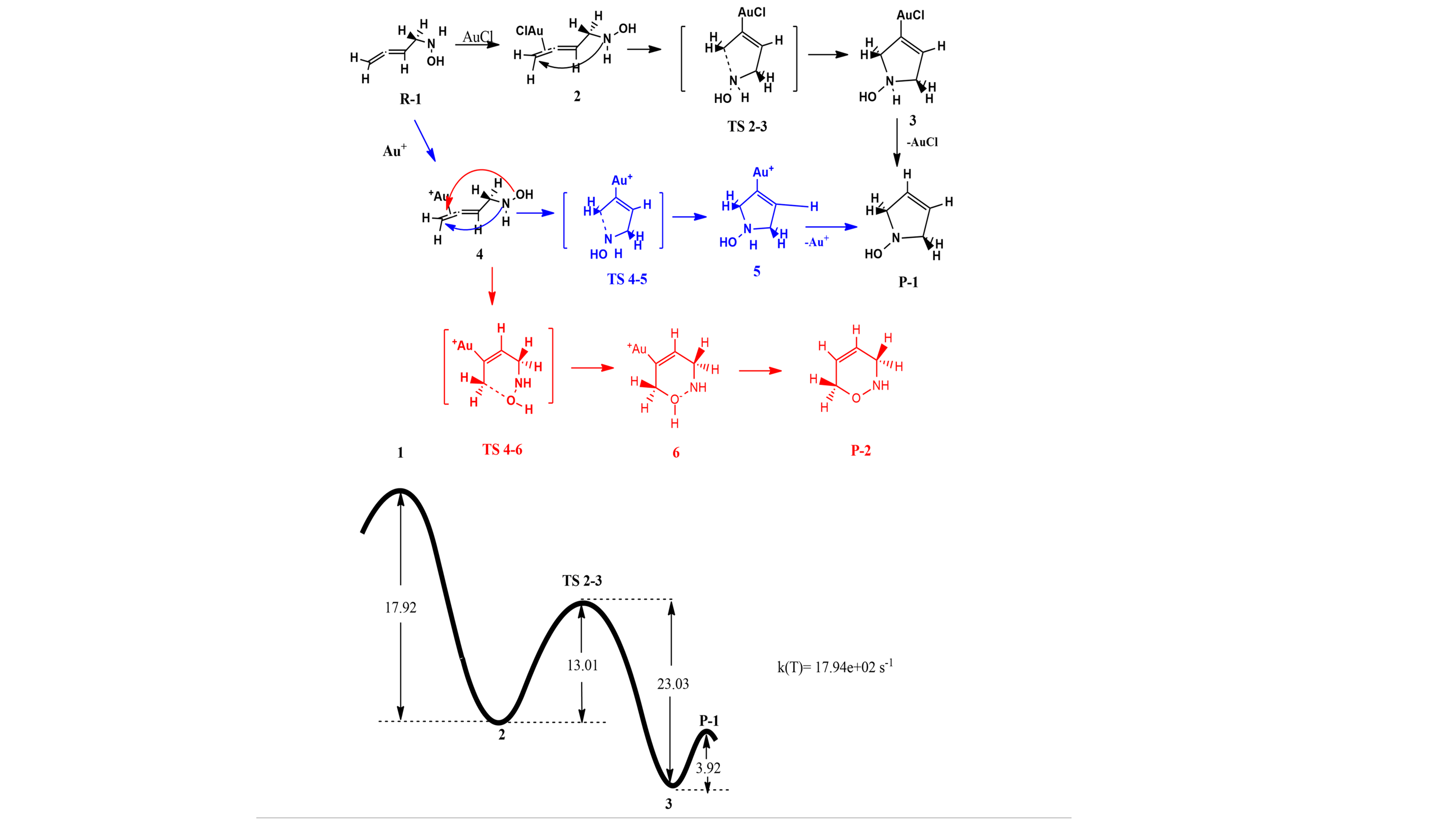
Scheme 1. Computational study of gold-catalyzed cycloisomerization of functionalized allenes.
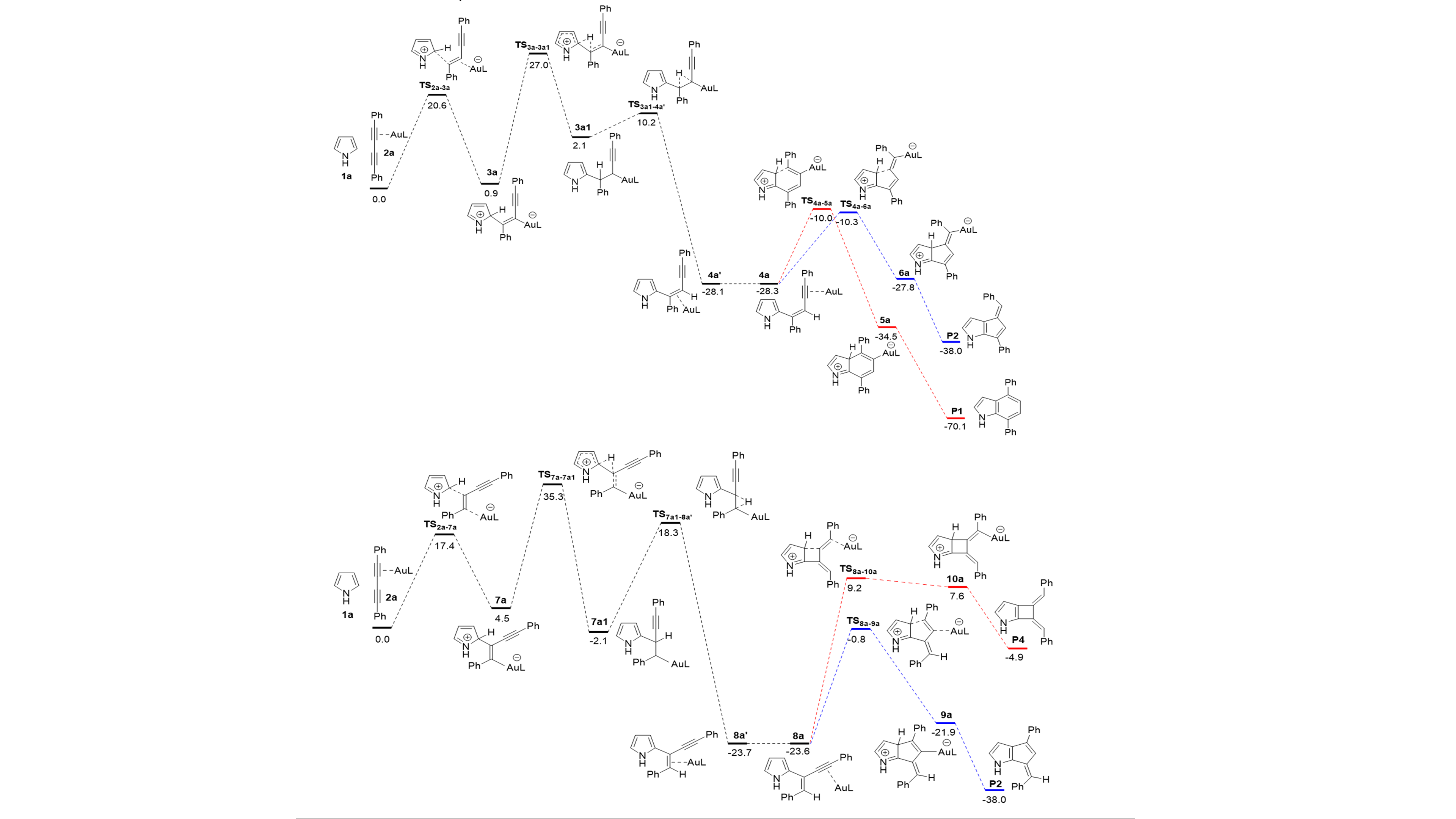
Scheme 2. Relative free energies (kcal/mol, 298 K and 1 atm) computed the alternative reaction pathways in the formal [4+2] reaction between pyrrole and diynes including protodeauration steps of key intermediates.
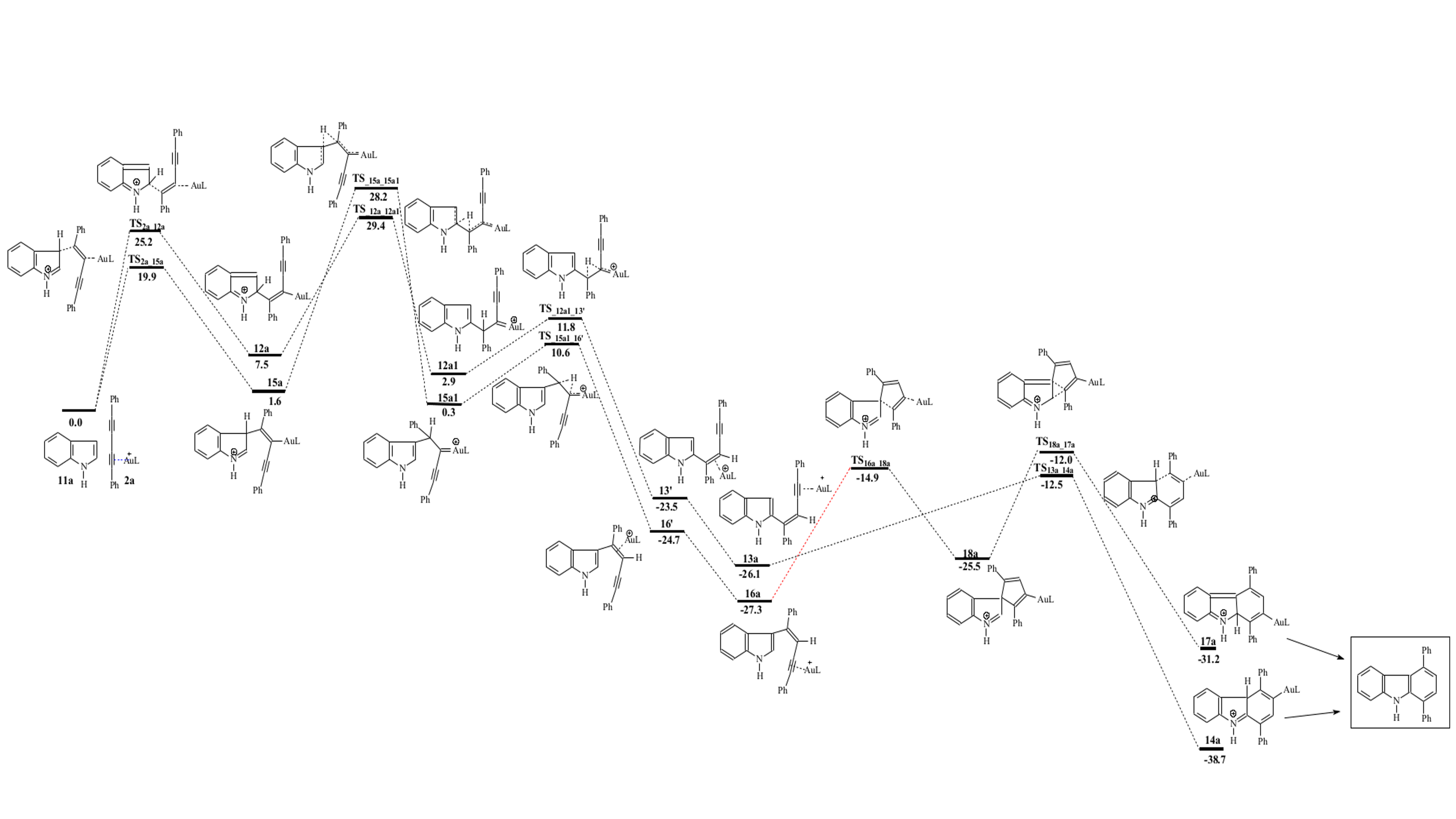
Scheme 3. Reactivity profile of indole with 1,3-diyne. Relative energies used compound 11a as the zero energy reference.