We are interested in exploring intramolecular and intermolecular C-H∙∙∙Y contacts and their possible contribution to the thermodynamic stability.
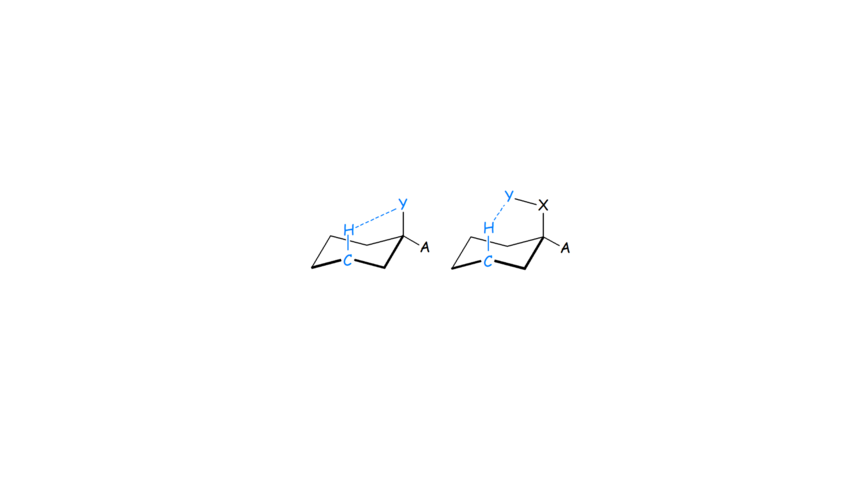
- We applied MP2 and various DFT functionals, NBO calculations and QTAIM analysis to propose for the first time that the C-Hax∙∙∙Yax-C contacts in the axial cyclohexane derivatives (Scheme 1) are not just Pauli repulsive but also include attractive dispersion interactions. They, may be characterized as improper or non-conventinal hydrogen-bonding interaction in certain cases (see Scheme 1, right part).
Scheme 1. C-Hax∙∙∙Yax contacts in cyclohexane derivatives.
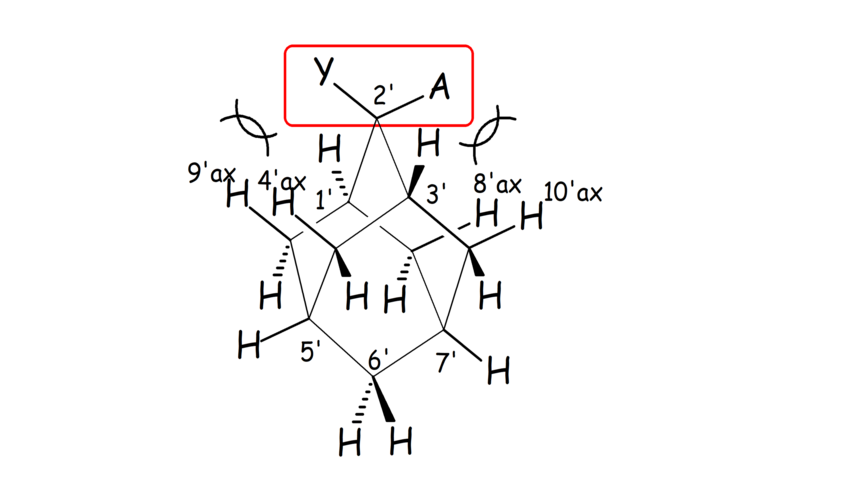
- We have published an unique finding for investigating the features of the 1H NMR spectrum of an axially substituted cyclohexane ring, through studying the rigid cyclohexane ring sub-units included in a suitable substituted adamantane derivative (Schemes 1, 2).
Scheme 2. In 2,2-disubstituted adamantanes each of the two substituents Y, A adopts an axial or equatorial orientation in a different cyclohexane ring sub-unit of adamantane.
- We have performed the full assignment of the 1H and 13C spectra numerous adamantane derivatives by applying 2D NMR spectra (Scheme 2). We also showed that the full assignment of the complicated 1H and 13C spectra of the adamantane derivatives can be analyzed and predicted in detail by applying GIAO calculations.
- We have linked the strength of cyclohexane bonded C-Hax∙∙∙Yax-C contacts using adamantane molecules as templates (Schemes 1, 2). A qualitative relation between chemical shift difference in 4',9'-axial and 4',9'-equatorial hydrogens and strength of these contacts was observed.
Weak intramolecular interactions and folding of transmembrane peptides
- Folding MD simulations were performed to study the a-helical stability of the structures between the influenza A transmembrane M2 protein (M2TM) 25-residues monomer and the 25-Ala (Ala25) peptide which is free of steric crowding from adjacent amino acid residues. This is in agreement with the MD simulations results of M2TM in trifluoroethanol as a membrane mimetic, using the AMBER99SB-STAR-ILDN force field, showing that the M2TM peptide did not form a single and stable α-helix compared to Ala25. Instead it appears to be dynamic in nature and quickly inter-converts between an ensemble of various folded helical structures.
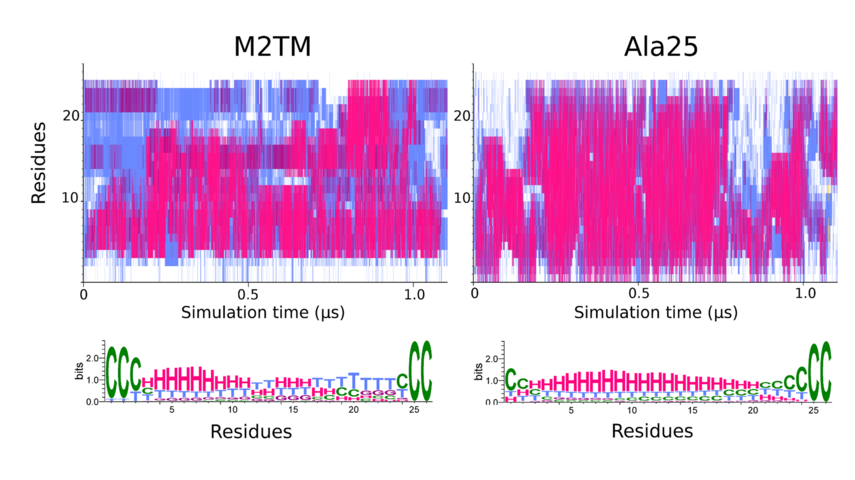
Figure 1. The two upper diagrams of the secondary structure analysis of the M2TM and Ala25 peptides depict the variation of conformation per-residue. STRIDE-derived secondary structure assignments as a function of simulation time. The color coding is red/magenta → α/310 helical structure, cyan → turns, white → coil. The lower panels show the weblogo-derived distributions corresponding to the STRIDE-derived assignments with H → α helix, G → 310 helix, T → turn, C → coil. The color coding is identical with the above except for coil which is depicted in green. For both peptides the terminal protection groups have been treated as discrete residues, with corresponding residue numbers of 0 and 26 respectively. The numbering refers to 1-25 aminoacids of the 25-residue M2TM peptide which are residues 22-46 in the 98-residues full M2 protein sequence.
A striking finding is that the residues with the highest thermal stability belong to the N-terminal part of the M2TM peptide, and not in Ala25 (Figure 1) (with Associate Professor N. Glykos, Biochemistry, Alexandroupolis, Professor Isaiah Arkin, Biochemistry, Jerusalem, Professor S. Scheiner, Chemistry, Utah).
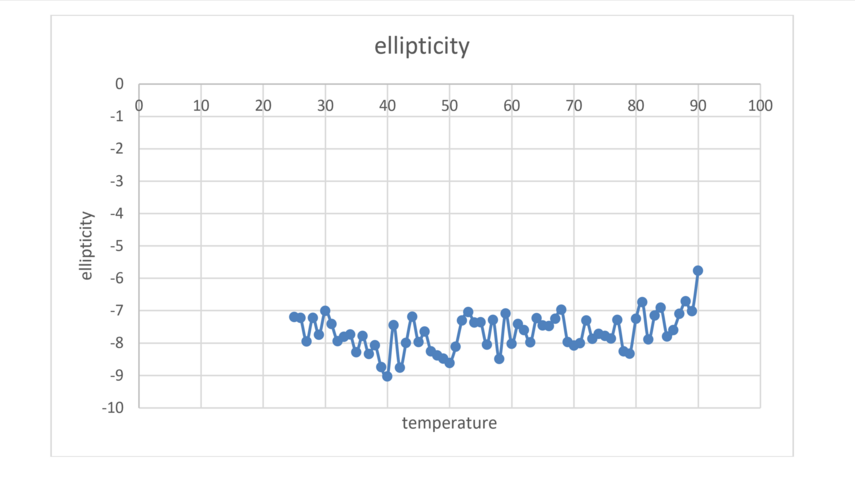
The MD results will be supported by experiments of the thermal stability of M2TM using CD measurements of the peptide helipticity (Figure 2).
Figure 2. Thermal melt results on M2 using CD.
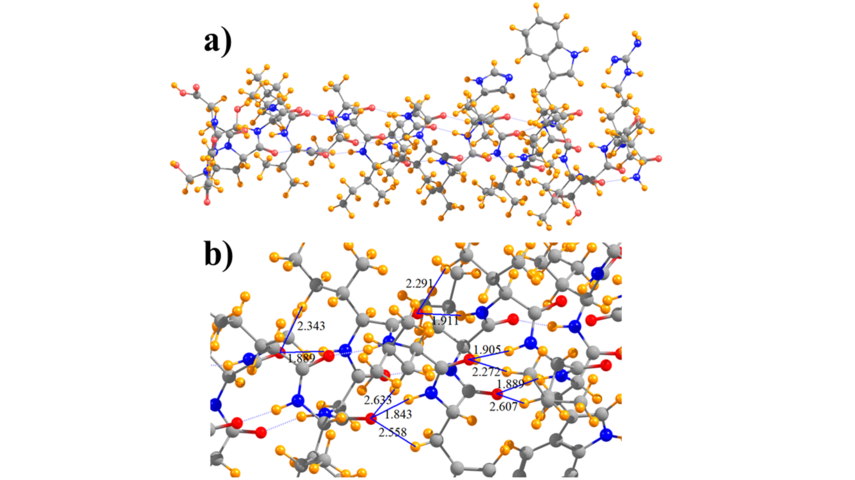
- DFT calculations were realized to analyze the forces in the M2TM which are likely to have a significant contribution to the folding from b-strand to the a-helix. On a technical level, calculations using D95(d,p) single point at a ONIOM (6-31G,3-21G) minimized geometry, in which the backbone is calculated with 6-31G and alkyl side chains with 3-21G, produced an energy differential for M2TM comparable with full D95(d,p) (Figure 3). The DFT calculations results revealed an extra stabilization for the folded α-helix in M2TM compared to Ala25 due to interactions that can't be described from a force field. Thus, natural bond orbital (NBO) and quantum theory of atoms in molecules (QTAIM) calculations were applied to investigate the relative contribution of N-H∙∙∙O as compared to C-H∙∙∙O hydrogen bonding interactions in the M2TM which included 17 lipophilic residues; 26 CH∙∙∙O interactions were identified, as compared to 22 NH∙∙∙O H-bonds. The calculations suggested that CH∙∙∙O hydrogen bonds, although individually weaker, have a cumulative effect that cannot be ignored and may contribute as much as half of the total interaction energy when compared to NH∙∙∙O to the stabilization of the folded α-helix in M2TM compared to Ala25 (Figure 4).
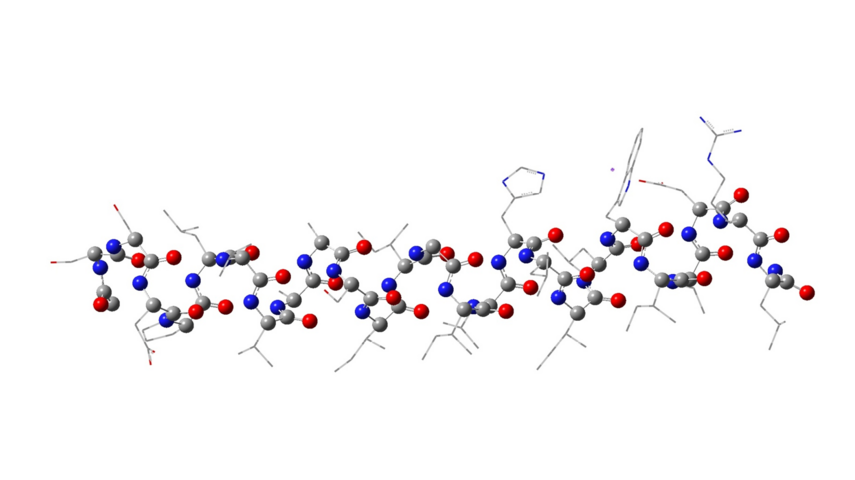
Figure 3. Structure of the peptide monomer of the M2TM influenza virus A using the ONIOM method and B3LYP/AM1 levels of theory.
Figure 4. Diagram of a) full model of M2TM and b) subsection that indicates selected NH···O and CH···O hydrogen bonds, and their lengths in Å.