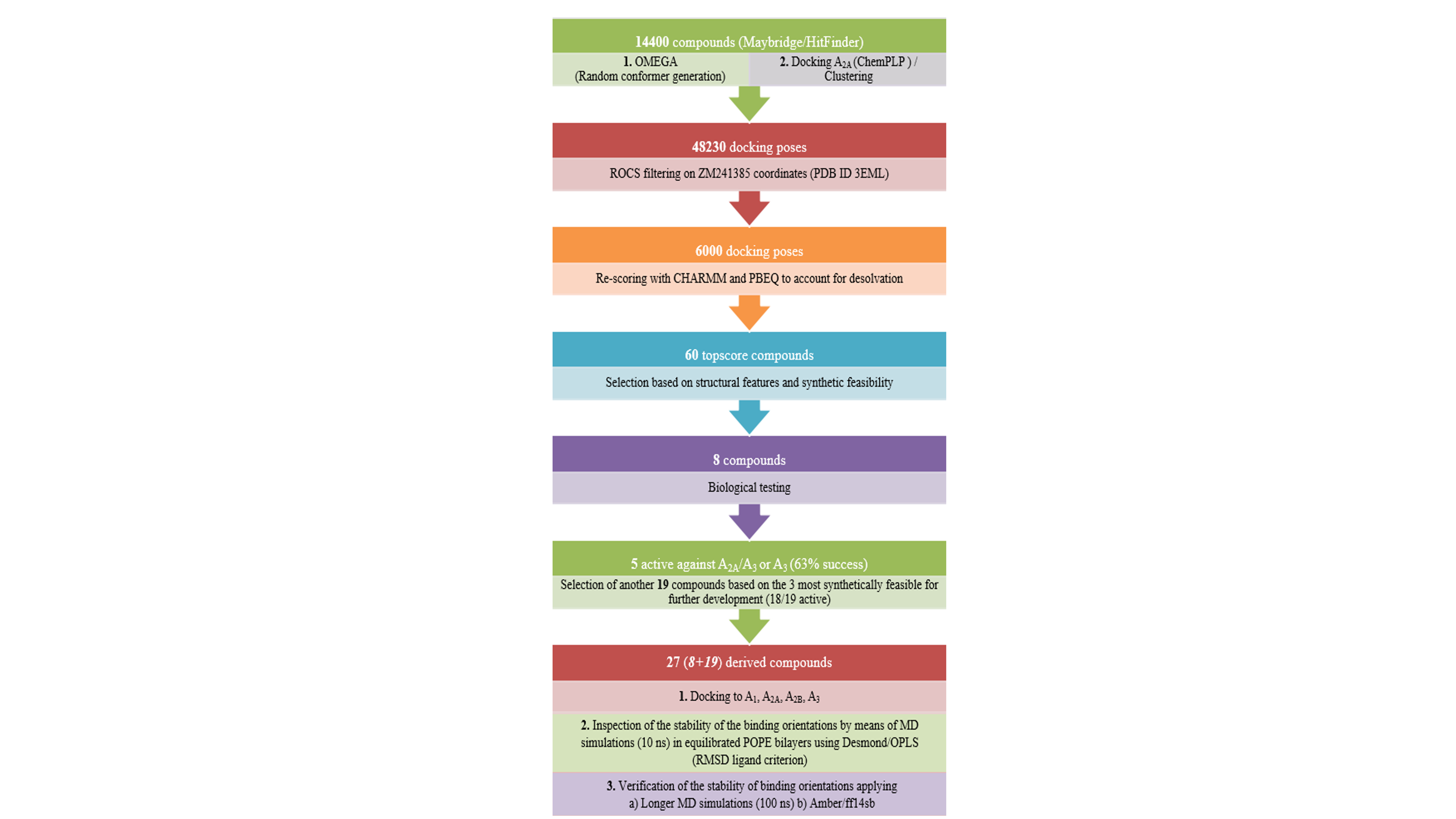
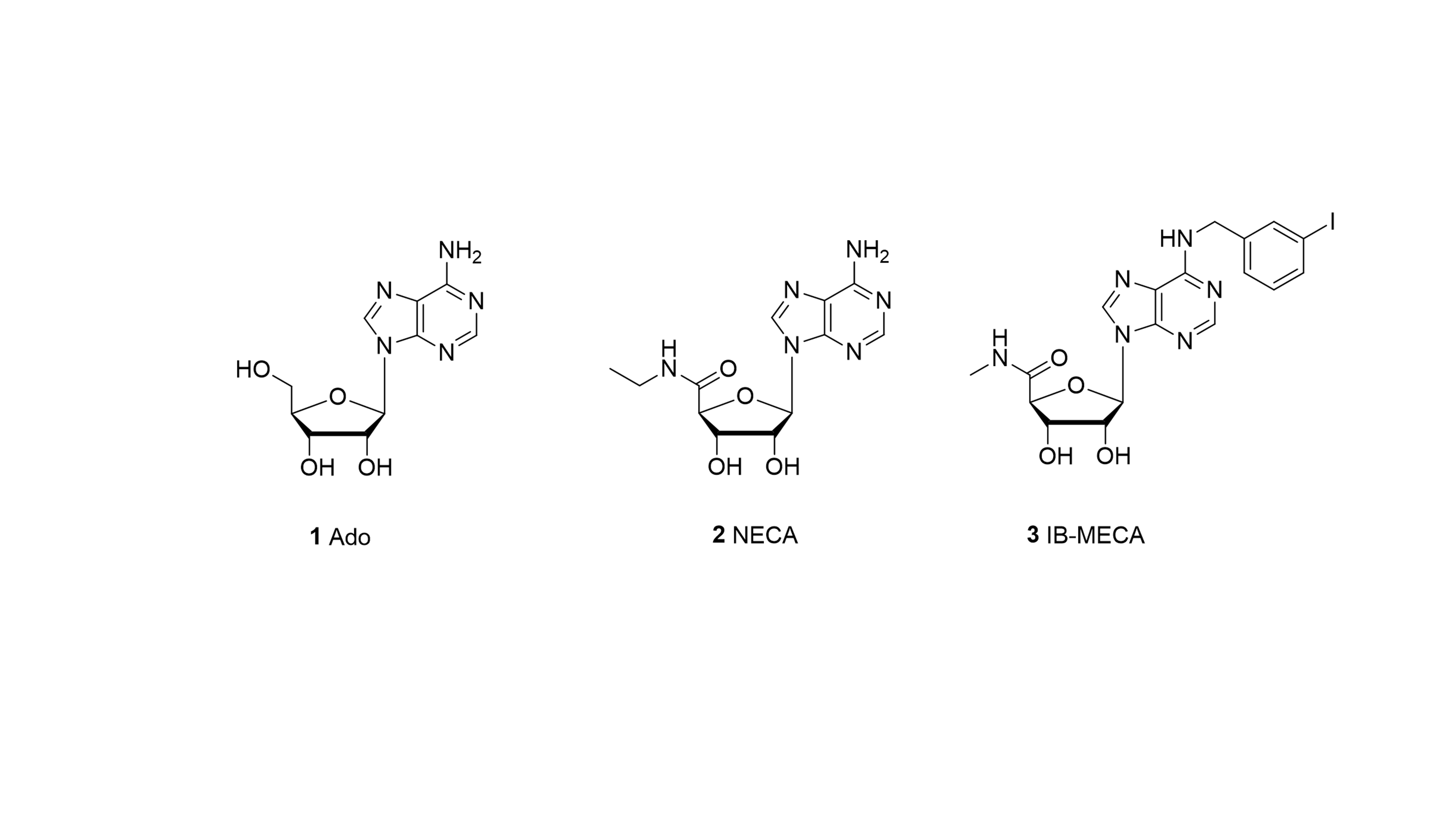
An intense effort is made by pharmaceutical and academic research laboratories to identify and develop selective antagonists for each adenosine receptor (AR) subtype as potential clinical candidates for "soft" treatment of various diseases. Crystal structures of subtypes A2A and A1ARs offer exciting opportunities for structure-based drug design.
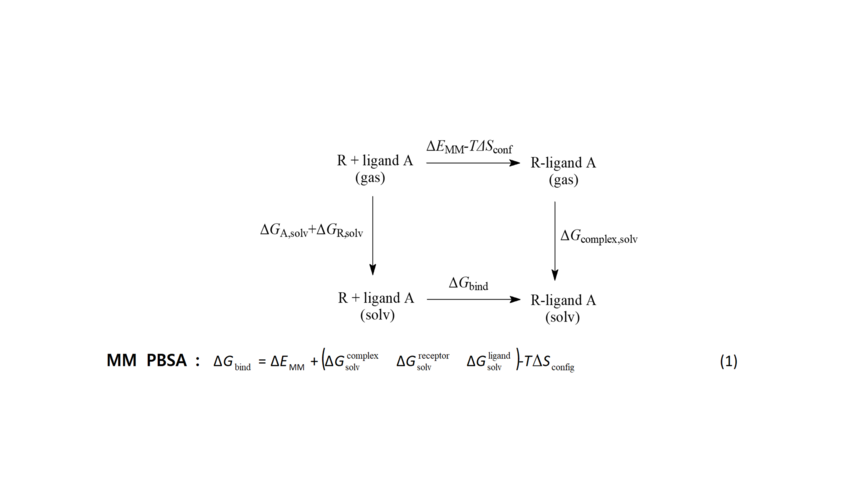
Scheme 2. The free energy for the formation of ligand A-receptor R complex can be calculated using the end-points of this thermodynamic cycle including the bound and unbound states of the ligand according to equation 1. The MM-PBSA model was applied. For the calculation of the free energies of solvation ΔGsolv ie, for transferring ligand, protein receptor and complex from the gas to the solution phase, the electrostatic component was calculated using the Poisson-Boltzmann equation and the non-polar component was calculated based on the surface of the cave formed by solute inside the solvent. ΔΕΜΜ is the molecular mechanics energy change between bound and unbound ligand. Entropy change is taken to be approximately zero.
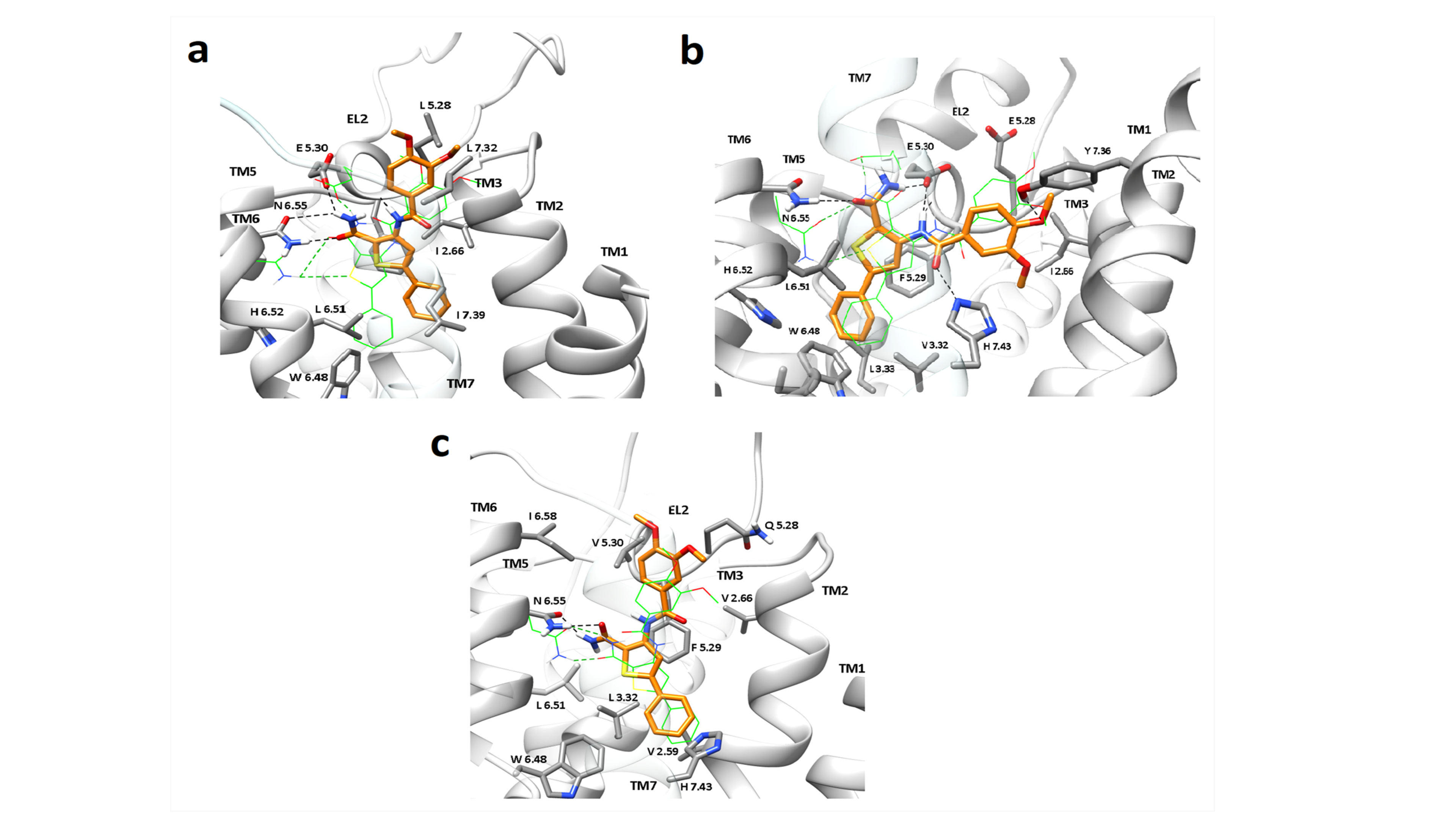
Figure 1. Predicted binding modes before and after 100 ns MD simulations for ligand 17 which binds A, A2A and A3 in the orthosteric binding site of (a) A1R, (b) A2AR and (c) A3R, resulting in stable complexes. Compound 17 can adopt a binding orientation inside A2AAR in which the amido group of thiophene ring is hydrogen-bonded to N(6.55) and E(5.30), and van der Waals interactions stabilize the ligand inside the binding cavity. The replacement of E(5.30) with V in A3R orthosteric cavity (panel c) retained the binding of compound 17, as a result of hydrogen-bonding to N(6.55) and additional favourable van der Waals interactions of its bulky lipophilic group in the vicinity of V(5.30). In A1AR, which has a broader binding cavity, the 5-aryl-thiophene ring is inclined and the 3-NHCOR substituent is directed towards TM2. Binding orientation of the ligand after the MD simulation is shown in yellow sticks and sidechains of some amino acids involved in ligand binding are displayed as gray sticks while the starting ligand and Ν(6.55)/Ε(5.30) side chain positions are shown in green wires. Hydrogen atoms are omitted except for those involved in hydrogen bond interactions, highlighted as black dashed lines.
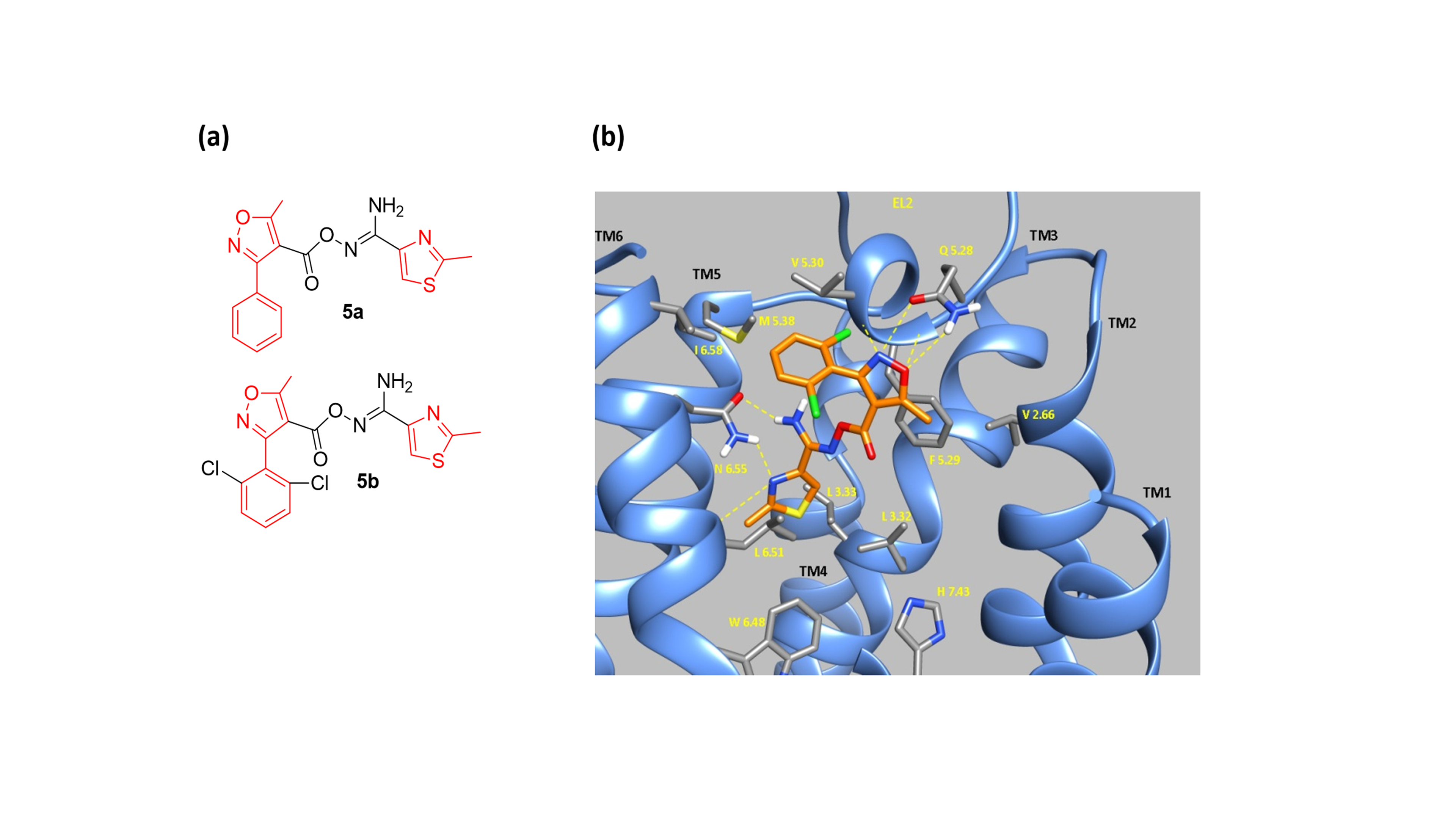
Figure 2. (a) Τhe 3-phenyloxazole 5b is a low micromolar A3R selective antagonist, and an analogue (but more active) of hit 5a. (b) MD screenshot of 5b inside A3AR orthosteric binding area.
A3AR is over-expressed in various tumor cells, compared to normal cells where it was found having low or no expression. Thus, A3AR and its signaling pathway is a promising drug target against cancer cell proliferation and for a number of other conditions like inflammatory diseases, including asthma and rheumatoid arthritis, and ischemic injury. Structures of subtypes A2A and A1ARs obtained using X-ray crystallography and the structure of A2A in complex with G-protein obtained using cryo-electron microscopy offer exciting opportunities for structure-based drug design. Currently there is no experimental structure for A3AR.
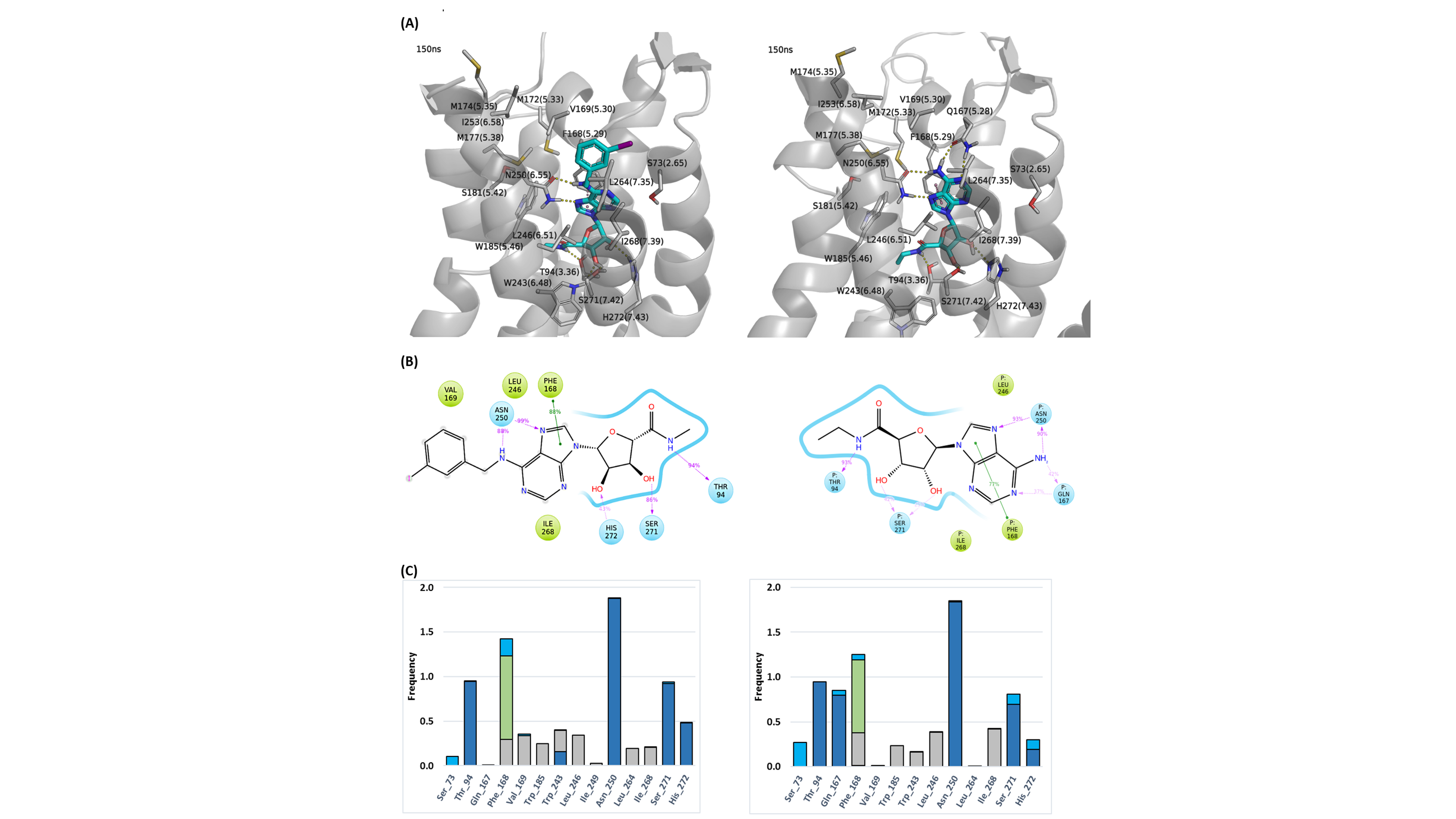
Figure 3. (A) Snapshot of IB-MECA (3) (left) and NECA (2) (right) inside the orthosteric binding area of WT A3R at 150 ns of the MD simulation, (B) 2D interaction diagram and (C) receptor-ligand interaction histogram for IB-MECA (3) (left) and NECA (2) (right) inside the orthosteric binding area of WT A3R for 0-150 ns of MD simulations. Bars are plotted for residues with interaction frequencies ≥ 0.2 for either IB-MECA (3) or NECA (2). Color scheme (A): Ligand=cyan sticks, receptor=white ribbon and sticks, H-bond interactions=yellow dashes, pi-pi interactions=pink dashes; Color scheme (B): polar surface/residues=blue, hydrophobic residues=green; Color scheme (C): H-bond interactions=dark blue, hydrophobic interactions=grey, pi-pi interactions=green, water-mediated interactions=light blue.